Vaccine News
New Report Confirms Pfizer Withheld “Efficacy” Announcement Until After 2020 Election

A new report done by Jeyanthi Kunadhasan, MD, FANZCA, and Ed Clark, MSE confirms that the Pfizer had it’s COVID-19 vaccines alleged “efficacy” data in October 2020, and decided to delay the announcement until November 9, 2020, after the 2020 U.S. Presidential election.
Pfizer claimed that less than a week after the U.S. presidential election, preliminary data from its coronavirus (COVID-19) vaccine indicate that the doses may be 90% effective at preventing COVID-19. Operation Warp Speed was launched by the Trump administration to identify vaccinations and treatments for COVID-19. President Donald Trump of the United States tweeted: “STOCK MARKET UP BIG, VACCINE COMING SOON.” Trump had frequently said during the campaign that a vaccination may be ready by election day. 90% EFFECTIVE REPORT. SUCH GREAT NEWS!” Since tracking the flu in 2003, scientists estimate that the vaccine is about 40% effective and that yearly doses are necessary.
Below is the report:
July 10, 2023 • by Jeyanthi Kunadhasan, MD, FANZCA, and Ed Clark, MSE – Team 3
Executive summary
- On July 21, 2020, the United States (U.S.) government, Pfizer, and BioNTech signed a contract, known as a Statement of Work (SOW), for the rapid development of and obtaining regulatory licensure for a COVID-19 vaccine by October 31, 2020, a few days prior to the November 3, 2020, U.S. Presidential election. Regulatory licensure was to be based on approval by the U.S. Food and Drug Administration (FDA) or authorization based on demonstration of efficacy. (https://www.hhs.gov/sites/default/files/pfizer-inc-covid-19-vaccine-contract.pdf)
- The July 2020 SOW states, “…if FDA approval or authorization was not issued by October 31, 2020…and Pfizer expects it will be unable to timely complete performance, then the Parties will discuss in good faith a contract modification to shift forward the estimated delivery schedule to reflect the difference in time period between October 31, 2020, and the date of actual regulatory approval or authorization.” In the SOW, Pfizer committed to notify the Government “of any event, risk, formal or informal FDA communication, or any other issue that would be reasonably expected to materially change the anticipated schedule by one week or more.” (https://www.hhs.gov/sites/default/files/pfizer-inc-covid-19-vaccine-contract.pdf, pp. 13-14.)
- On the date of Pfizer Protocol Amendment 9, October 29, 2020, Pfizer already had more than 62 evaluable efficacy cases diagnosed, as required in that exact amendment. (“125742_S1_M5_5351_c4591001-interim-mth6-protocol.pdf,” https://phmpt.org/wp-content/uploads/2022/03/125742_S1_M5_5351_c4591001-interim-mth6-protocol.pdf, p. 935)
- On November 9, 2020, Pfizer announced that its BNT162b2 vaccine candidate was >90% effective based on a November 8, 2020, interim analysis of partial data from its Phase 3 clinical trial. (“Pfizer and BioNTech Announce Vaccine Candidate Against COVID-19 Achieved Success in First Interim Analysis from Phase 3 Study.” Retrieved June 19, 2023, from https://www.businesswire.com/news/home/20201109005539/en/.)
- Despite Pfizer not making its vaccine efficacy announcement until November 9, 2020 (https://www.keionline.org/misc-docs/FOIA/DoD-Pfizer-Contract-W15QKN21C0012-22Dec2020.pdf, p. 3), more than one week after the October 31, 2020, SOW commitment date, there is no evidence that Pfizer notified the Government, as required, of a greater than one-week change in schedule. (Department of Defense (DOD)-Pfizer Contract W15QKN21C0012 (includes Modification 310). December 22, 2020. (n.d.). Retrieved from https://www.keionline.org/misc-docs/FOIA/DoD-Pfizer-Contract-W15QKN21C0012-22Dec2020.pdf, p. 3.) (“Pfizer and BioNTech Announce Vaccine Candidate Against COVID-19 Achieved Success in First Interim Analysis from Phase 3 Study.” Retrieved June 19, 2023, from https://www.businesswire.com/news/home/20201109005539/en/.)
- On November 18, 2020, Pfizer reported 95% effectiveness based on analysis of a larger dataset that included 170 patients (162 in the placebo group and 8 in the vaccinated group). Based on the strength of that data, Pfizer formally requested Emergency Use Authorization (EUA) from the US Food and Drug Administration.” (Department of Defense (DOD)-Pfizer Contract W15QKN21C0012 (includes Modification 310). December 22, 2020. (n.d.). Retrieved from https://www.keionline.org/misc-docs/FOIA/DoD-Pfizer-Contract-W15QKN21C0012-22Dec2020.pdf, p. 3.)
- The FDA then authorized the EUA for Pfizer’s COVID-19 vaccine on December 11, 2020. (https://www.fda.gov/media/144416/download)
- From the analysis of the 170 patients found in the FDA-released Pfizer clinical trial documents (https://phmpt.org/pfizer-16-plus-documents/), there is evidence that Pfizer had the data needed for its interim analysis of the “evaluable efficacy” in October 2020, when the various accrual thresholds of evaluable efficacy cases (i.e., the total number of COVID-19-positive patients that Pfizer counted to prove “efficacy”) were reached. Therefore, an announcement of vaccine efficacy could have been made in October 2020, before the U.S. 2020 presidential election. When you read on, you will likely have the same question we do: Was this unnecessarily delayed announcement an effort to negatively impact one candidate’s chances of winning the U.S. Presidential election?
On July 21, 20202, the U.S. government and Pfizer/BioNTech signed a Statement of Work (SOW) for COVID-19 vaccine development. (Technical Direction Letter for Medical CRBN Defense Consortium (MCDC), Request for Prototype Proposals (RPP) 20-11, Objective PRE-20-11 for “COVID-19 Pandemic — Large Scale Vaccine Manufacturing Demonstration’ (Pfizer, Inc.).” HHS.gov, 21 July 2020. Retrieved from https://www.hhs.gov/sites/default/files/pfizer-inc-covid-19-vaccine-contract.pdf.) The goal of the prototype program was to “…rapidly develop and obtain regulatory licensure for a COVID-19 vaccine for use in adults ≥18 years of age.” According to the SOW, the goal was to have an FDA-approved or FDA-authorized vaccine ready for administration in the United States by October 31, 2020.

(Technical Direction Letter for Medical CRBN Defense Consortium (MCDC), Request for Prototype Proposals (RPP) 20-11, Objective PRE-20-11 for “COVID-19 Pandemic — Large Scale Vaccine Manufacturing Demonstration’ (Pfizer, Inc.).” HHS.gov, 21 July 2020. Retrieved from https://www.hhs.gov/sites/default/files/pfizer-inc-covid-19-vaccine-contract.pdf, p. 3.)
The scope of the prototype project (https://www.hhs.gov/sites/default/files/pfizer-inc-covid-19-vaccine-contract.pdf, p. 10) was “…the demonstration by Pfizer of the supply and logistics capability to manufacture and distribute to the Government of 100M doses of a novel mRNA-based vaccine that has received FDA approval or authorization based on demonstration of efficacy (hereafter FDA-approved or authorized). “
Due to the perceived need for rapid vaccine development, the Pfizer/BioNTech trial for this vaccine, U.S. study C4591001, was designed as a single, multistage, and multiphase trial. As such, the trial protocol would be amended continuously whilst the clinical trial was underway. A trial was run as outlined in the protocol, and major modifications to the protocol are reflected in formal protocol amendments.
Phases 1 and 2 of the vaccine candidates (i.e., the possible vaccine formulations) had been ongoing since May 2020. With the rapidly evolving pandemic situation, there was a perceived need to demonstrate vaccine efficacy as soon as possible. Protocol Amendment 4 (June 30, 2020) had the first interim analysis plan. (“125742_S1_M5_5351_c4591001-interim-mth6-protocol.pdf,” https://www.phmpt.org/wp-content/uploads/2022/03/125742_S1_M5_5351_c4591001-interim-mth6-protocol.pdf, p. 1792.) There were to be four interim analyses performed by an unblinded statistical team after accrual of 32, 62, 92, and 120 COVID-19 cases that met the trial’s primary endpoint (i.e., the “evaluable efficacy population,” which will be explained in detail). The goal was a total of 164 “evaluable efficacy” cases. This is mentioned on page 7 of the SOW, stating that the “…study currently is being amended to incorporate a pivotal efficacy study design.”
Protocol Amendment 5, dated July 24, 2020, clarified that a single vaccine candidate, BNT162b2 at a dose of 30 μg administered as two doses 21 days apart, would be studied in Phase 2/3 of the trial. (“125742_S1_M5_5351_c4591001-interim-mth6-protocol.pdf,” https://www.phmpt.org/wp-content/uploads/2022/03/125742_S1_M5_5351_c4591001-interim-mth6-protocol.pdf,. p. 1539) The official start date for Phase 3 was July 27, 2020, six days after the SOW was signed. From pages 13 and 14 of the SOW, “…if FDA approval or authorization was not issued by October 31, 2020…and Pfizer expects it will be unable to timely complete performance, then the Parties will discuss in good faith a contract modification to shift forward the estimated delivery schedule to reflect the difference in time period between October 31, 2020 and the date of actual regulatory approval or authorization.” (https://www.hhs.gov/sites/default/files/pfizer-inc-covid-19-vaccine-contract.pdf) Also, in addition to regular reporting requirements, during the period of performance, Pfizer committed to notify the government of any event, risk, formal or informal FDA communication, or any other issue that would be reasonably expected to materially change the anticipated schedule by ONE WEEK or more. (pp. 13-14, bold and capitalization added.)
No COVID-19 vaccine was approved by October 2020. A contract modification simply stated, “On November 9, 2020, Pfizer announced that BNT162b2 was >90% effective based on interim analysis of partial data from its Phase 3 clinical trial. On November 18, 2020, Pfizer reported 95% effectiveness based on analysis of a larger dataset that included 170 volunteers (162 in the placebo group and 8 in the vaccinated group). Based on the strength of this data, Pfizer formally requested Emergency Use Authorization (EUA) from the US Food and Drug Administration.” (Department of Defense (DOD)-Pfizer Contract W15QKN21C0012 (includes Modification 310). December 22, 2020. (n.d.). Retrieved from https://www.keionline.org/misc-docs/FOIA/DoD-Pfizer-Contract-W15QKN21C0012-22Dec2020.pdf, p. 3.)

(DoD-Pfizer Contract W15QKN21C0012 (includes Mod 310). December 22, 2020. (n.d.). Retrieved from https://www.keionline.org/misc-docs/FOIA/DoD-Pfizer-Contract-W15QKN21C0012-22Dec2020.pdf, p. 3.)
Previous reporting shows that the 170 patients formed the basis of the EUA application and approval. (Kunadhasan, J., Clark, E., & Flowers, C. (October 21, 2022). “Report 42: Pfizer’s EUA Granted Based on Fewer Than 0.4% of Clinical Trial Participants. FDA Ignored Disqualifying Protocol Deviations to Grant EUA.” Retrieved from https://dailyclout.io/report-41-the-170-clinical-trial-participants-who-changed-the-world-pfizer-ignored-protocol-deviations-to-obtain-emergency-use-authorization-for-its-covid-19-mrna-vaccine/.)
What is the evaluable efficacy population?
The evaluable efficacy population is the primary endpoint of Pfizer’s trial. An endpoint is a measurable outcome used to determine whether a drug under investigation is beneficial or not. To qualify to be part of the evaluable efficacy population, all eligible, randomized participants must:
- Receive all vaccination(s) as randomized within the predefined window.
- Have no evidence of COVID infection prior to seven days after the second dose of the vaccine.
- Have the efficacy measurement (i.e., the test confirming symptomatic COVID-19 infection) only after seven days following the second vaccine dose.
- Have no other major protocol deviations as determined by the clinician.
A major protocol deviation excluded a participant from the evaluable efficacy population from the date that it occurred through the participant’s remaining follow-up. Vaccine efficacy is measured by calculating the risk of disease among the vaccinated and placebo groups and determining the percentage reduction in disease between the two groups.
These COVID cases among these 170 patients were the basis of the claim of “95% effective.” We had already published that, in the 170 patients, five had dosing interval irregularities, one did not receive the correct dose of the investigational product, and another received a blood product within 60 days (a confounding event for infection), all of which should have disqualified them from being part of the evaluable efficacy population. Two others had been withdrawn from the trial prior to issuance of the Emergency Use Authorization (EUA). These disqualified patients would have brought the final number of cases to fewer than the 164 patient threshold, thus bringing into question if an EUA application could have been made, much less approved. To date there is no list available as to what constitutes a “major” protocol deviation that clinicians can review to discern if certain protocol deviations, presumably now reincluded into the efficacy analysis, have a significant effect on treatment efficacy.
When were the 170 evaluable efficacy cases diagnosed?
In our analysis, we have made no attempt to critique the study design and endpoints chosen. We are only assessing if the protocol was followed and if public statements by Pfizer correlate with what we found in the FDA-released Pfizer documents [https://phmpt.org/pfizer-16-plus-documents/].
In this report, we will focus on the timing of the accrual of the 170 evaluable efficacy patients.
First, one must understand what the public was led to believe.
The Phase 3 portion of the trial started on July 27, 2020. Half of the Phase 3 trial participants were recruited by September 1, 2020.
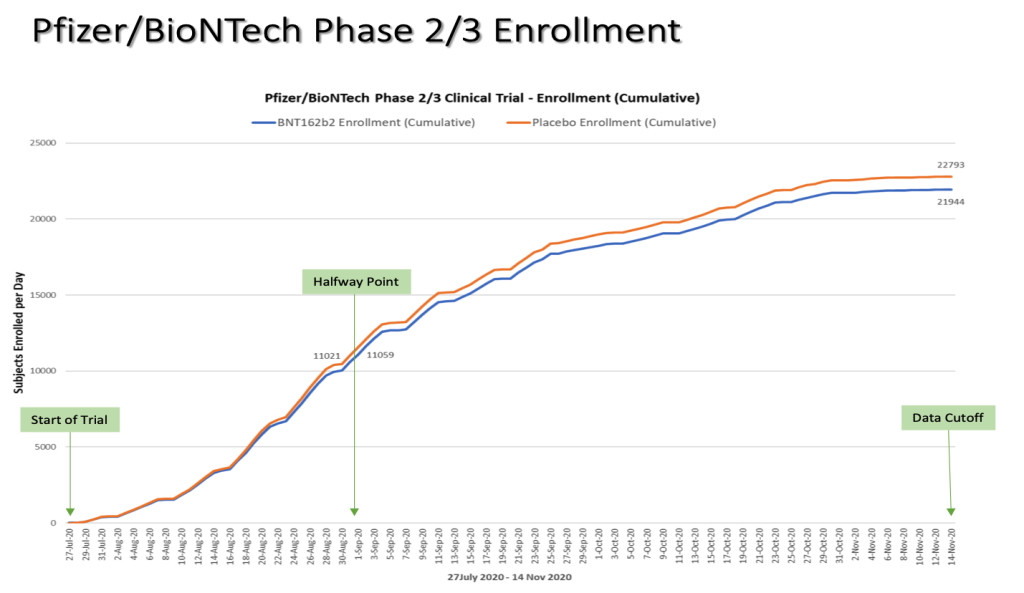
Chart by Ed Clark
On October 29, 2020, a formal protocol amendment (the ninth) was made and clarified that interim analyses would be conducted after accrual of at least 62, 92, and 120 cases. (“125742_S1_M5_5351_c4591001-interim-mth6-protocol.pdf,” https://phmpt.org/wp-content/uploads/2022/03/125742_S1_M5_5351_c4591001-interim-mth6-protocol.pdf, p. 935) Given that there was an October 29 protocol amendment made stating that an interim analysis would be “…conducted after accrual of at least 62 cases…,” fewer than 62 cases must have diagnosed by that date.

(125742_S1_M5_5351_c4591001-interim-mth6-protocol.pdf. Retrieved from https://phmpt.org/wp-content/uploads/2022/03/125742_S1_M5_5351_c4591001-interim-mth6-protocol.pdf, p. 935.)
What the public was led to believe without access to FDA-released Pfizer documents and data
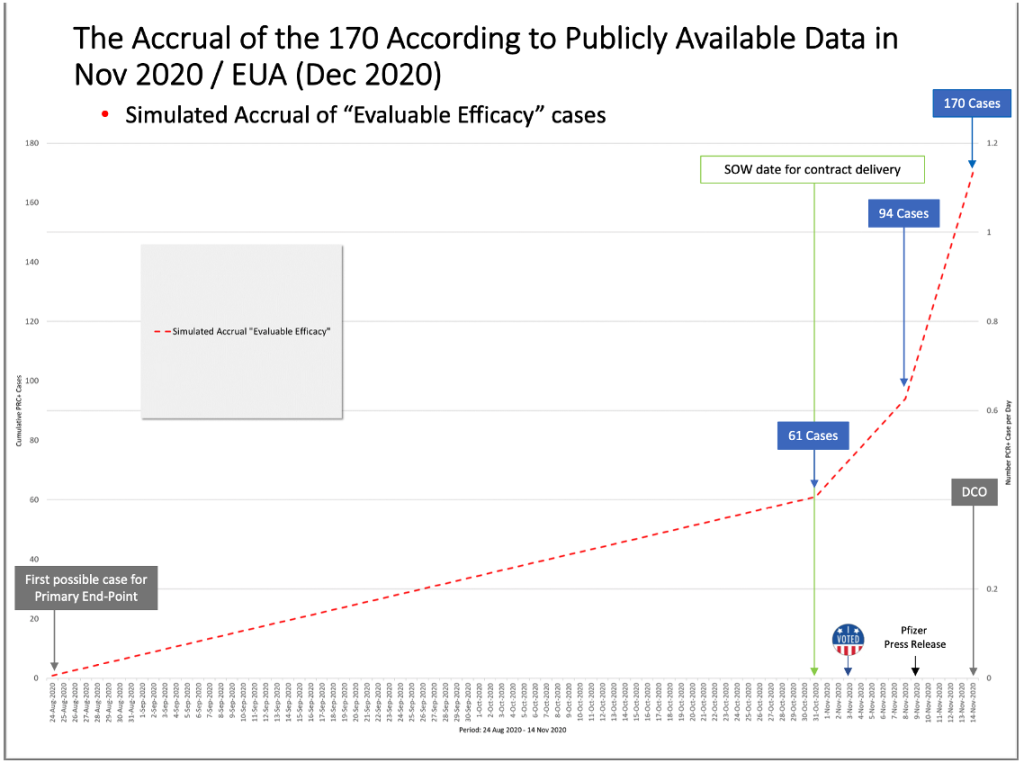
DCO = Data Cut-Off
Based on the October 29, 2020, protocol amendment stating that an interim analysis would be done when there were at least 62 cases accrued, and the lack of vaccine approval by October 31, 2020, fewer than the 62 cases needed for Interim Analysis 2 must have accumulated by the end of October 2020. It is unknown how many “evaluable efficacy” cases were actually confirmed by Pfizer by that date which enabled the DoD and Pfizer/BioNTech contract to be modified in “good faith.”
Yet, by the month of October 2020, more than half of the trial participants had already been recruited and completed the two-dose vaccination schedule, thus reaching the eligible timeframe to be included in the primary endpoint evaluable efficacy population. With a two-dose interval of 21 days and seven days post Dose 2, the first possible date to be eligible for the primary endpoint was August 24, 2020; however, the apparent rate of accrual of eligible cases from the August 24, 2020, to October 31, 2020, a period of 67 days, was only about 0.9 cases per day, or 61 cases over 67 days. (Note that the accrual rate must be derived from available documents and Pfizer’s statements since Pfizer and the DoD did not announce the number of evaluable efficacy cases through the end of October.)
After evaluating 94 confirmed cases of COVID-19 in trial participants in its first interim analysis conducted on November 8, 2020, Pfizer announced in a press release on November 9, 2020, that the company’s vaccine candidate was found to be more than 90% effective at preventing COVID-19. (“Pfizer and BioNTech Announce Vaccine Candidate Against COVID-19 Achieved Success in First Interim Analysis from Phase 3 Study.” Retrieved June 19, 2023, from https://www.businesswire.com/news/home/20201109005539/en/.)
Hence, from October 31, 2020, until November 8, 2020, there was a jump in cases diagnosed from 61 to 94, a rate of about four cases a day. Then, an even bigger jump of cases occurred from November 8, 2020, through November 14, 2020 (the data cutoff date), from 94 cases to the final 170 cases or about 12 cases diagnosed per day.
In analyzing this, we also noted the inconsistencies in Pfizer’s public statements and the dates noted in official documentation. For example, on page 17 of the EUA documentation authorizing the vaccine it stated, “The date for data cut-off for the first interim analysis for efficacy was November 4, 2020, when a total of 94 confirmed COVID-19 cases were accrued.” (Gruber, M. F. (December 11, 2020). EUA Decision Memorandum. Retrieved from https://www.fda.gov/media/144416/download.) This is inconsistent with an article in USA Today that stated, “Pfizer lacked access to its trial data until after Election Day and could not have known or released the results prior to that. Albert Bourla, Pfizer’s CEO, told Axios that the data came in on Nov. 5 or 6, after Election Day on Nov. 3.” (Caldera, C. (2020, November 18). Fact check: Pfizer received COVID-19 vaccine data after Election Day, released within days. Retrieved June 19, 2023, from https://www.usatoday.com/story/news/factcheck/2020/11/18/fact-check-pfizer-received-covid-vaccine-data-after-election-day/6267242002/.) This begs the question as to how there could been the required “good faith” in fulfilling the contract by October 31, 2020, with a process that appears to have been set up to look into the data only after November 3, 2020, the date of the U.S. Presidential election.
When were the 170 evaluable efficacy cases diagnosed?
As we have found the 170 patients upon which the efficacy criterion for the EUA approval were based, we have also been able to plot the date of the accrual of the 170 patients. We were able to obtain the diagnosis date by combing through pages 1059 to 2506 of the Pfizer narrative documents released by the FDA. (125742_S1_M5_5351_c4591001-fa-interim-narrative-sensitive.pdf. (2022, July 1). Retrieved from https://pdata0916.s3.us-east-2.amazonaws.com/pdocs/070122/125742_S1_M5_5351_c4591001-fa-interim-narrative-sensitive.pdf.) One caveat is that, at times, the diagnosis date was not explicitly stated in the documents, in which case the swab date was used as a proxy date. The narrative document was finalized on December 4, 2020. In another document, finalized on April 1, 2021, one is able to use the list of 170 patients to get the dates that the central lab swab was taken by going through pages 226 to 416. (125742_S1_M5_5351_c4591001 interim mth6 lab measurements sensitive.pdf. (2022, July 1). Retrieved from https://www.phmpt.org/wp-content/uploads/2022/07/125742_S1_M5_5351_c4591001-interim-mth6-lab-measurements-sensitive.pdf, pp. 1059-2506.) In another set of documents dated June 2, 2021, one can find the dates when the 170 patients’ central swab tests returned a positive result. The swabs were presumably processed only in Pfizer’s lab in Pearl River, New York, since it is the company’s “primary location for…global Vaccine Research and Development.” (https://www.pfizer.com/science/centers) (125742_S6_M5_5351_c4591001 sequencing listings.pdf. (2023, January 3). Retrieved from https://www.phmpt.org/wp-content/uploads/2023/01/125742_S6_M5_5351_c4591001-sequencing-listings.pdf.)
Publicly available Pfizer documentation causes confusion as to when precisely patients were diagnosed with COVID-19. If we look at Pfizer’s protocol, the diagnosis of cases was supposed to happen based on the results of a central laboratory test. There are certain conditions when the test results could be based on a local test.

(125742_S1_M5_5351_c4591001-interim-mth6-protocol.pdf. (n.d.). Retrieved from https://www.phmpt.org/wp-content/uploads/2022/03/125742_S1_M5_5351_c4591001-interim-mth6-protocol.pdf page 1591.)
Let us review the example of a patient, ID 10091128.
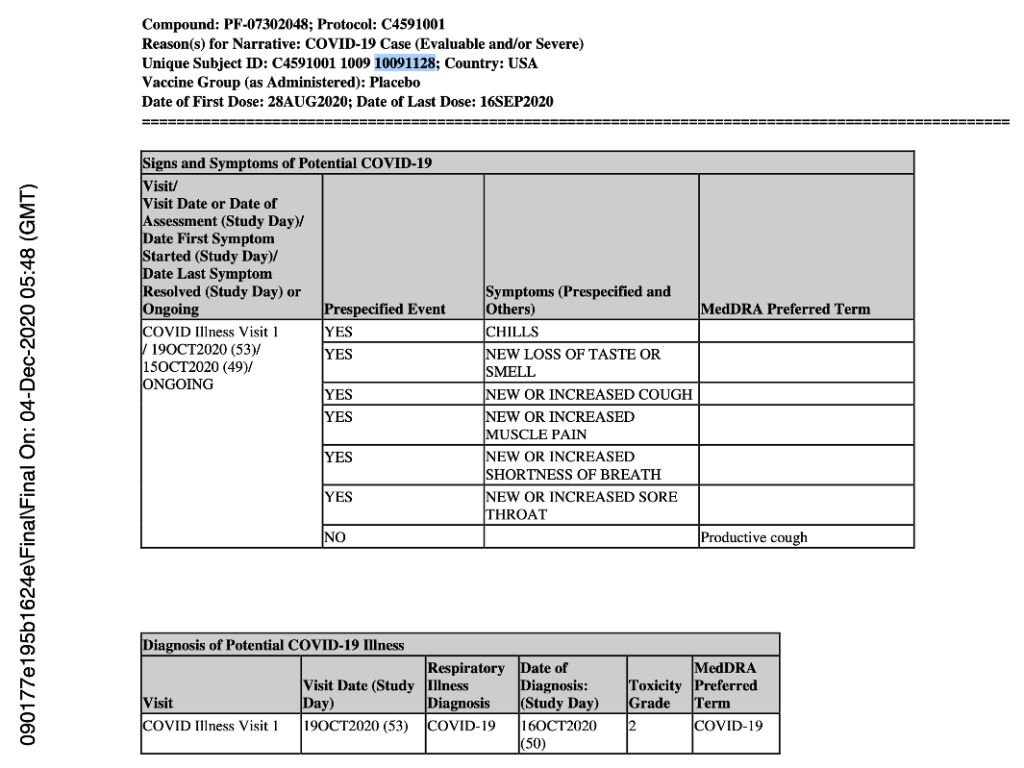
(125742_S1_M5_5351_c4591001-fa-interim-narrative-sensitive.pdf. (2022, July 1). Retrieved from https://pdata0916.s3.us-east-2.amazonaws.com/pdocs/070122/125742_S1_M5_5351_c4591001-fa-interim-narrative-sensitive.pdf, p. 1163.)
As you can see, the date of diagnosis is explicitly stated as October 16, 2020, even though the central lab test was done on October 19, 2020, as shown in the document finalized on December 4, 2020 (presumably a document used to apply for EUA, which was approved just one week later on December 11, 2020). (https://pdata0916.s3.us-east-2.amazonaws.com/pdocs/070122/125742_S1_M5_5351_c4591001-fa-interim-narrative-sensitive.pdf) However, for that same patient, in a third document finalized in June 2021, the date of the central lab test is October 20, 2020. If taking that as a date of diagnosis for this patient, it would now be four days apart from the previous date of October 16, 2020.
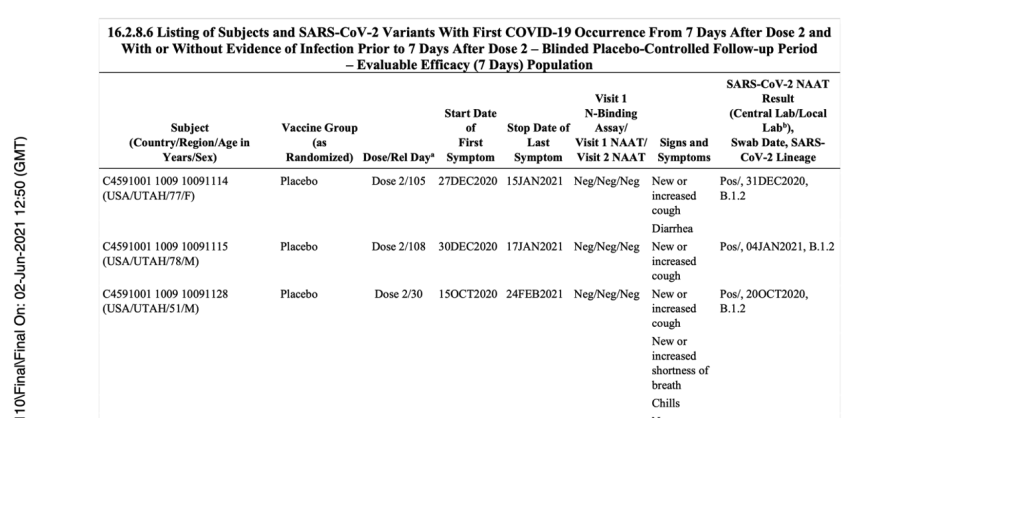
(125742_S6_M5_5351_c4591001 sequencing listings.pdf. (2023, January 3). Retrieved from https://phmpt.org/wp-content/uploads/2023/01/125742_S6_M5_5351_c4591001-sequencing-listings.pdf, p. 16.)
In December 2022, we published this approximate accrual of when the 170 subjects were diagnosed based on the interim narrative documents, explicitly stating date of diagnosis in the Pfizer documents finalized on December 4, 2020. (Kunadhasan, Jeyanthi. (2022, December 8). “170 patients that changed everything.” spectator.com.au. Retrieved June 19, 2023, from https://www.spectator.com.au. Kunadhasan, Jeyanthi. “‘170 Patients That Changed Everything’ – Spectator: Australia.” DailyClout, 8 Dec. 2022, https://dailyclout.io/170-patients-that-changed-everything-spectator-australia/.)
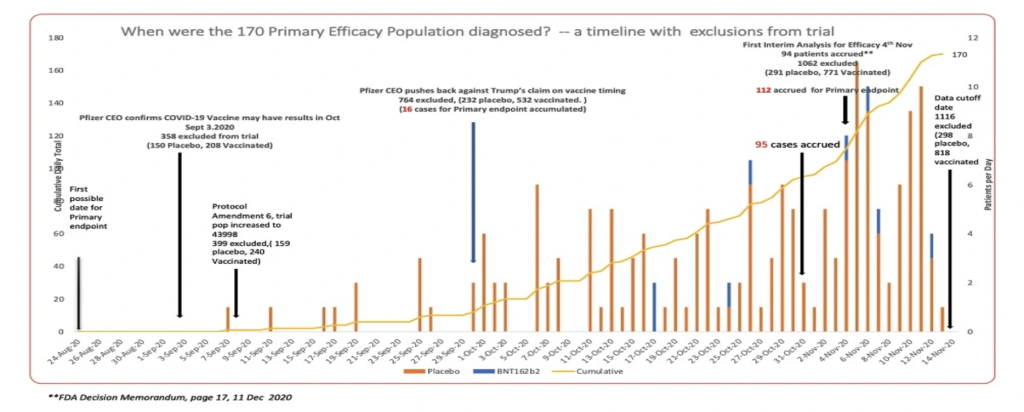
(Note the disproportionate excluded vaccinated patients versus placebo patients in the run up to the data cut-off date. This requires separate analysis and will not be covered in this report.)
We have analyzed the differing accruals of the 170 population based on documents finalized in December 2020 (https://pdata0916.s3.us-east-2.amazonaws.com/pdocs/070122/125742_S1_M5_5351_c4591001-fa-interim-narrative-sensitive.pdf) and June 2021 (https://www.phmpt.org/wp-content/uploads/2023/01/125742_S6_M5_5351_c4591001-sequencing-listings.pdf) and further cross-checked with data from the FDA-released Pfizer XPT database files.
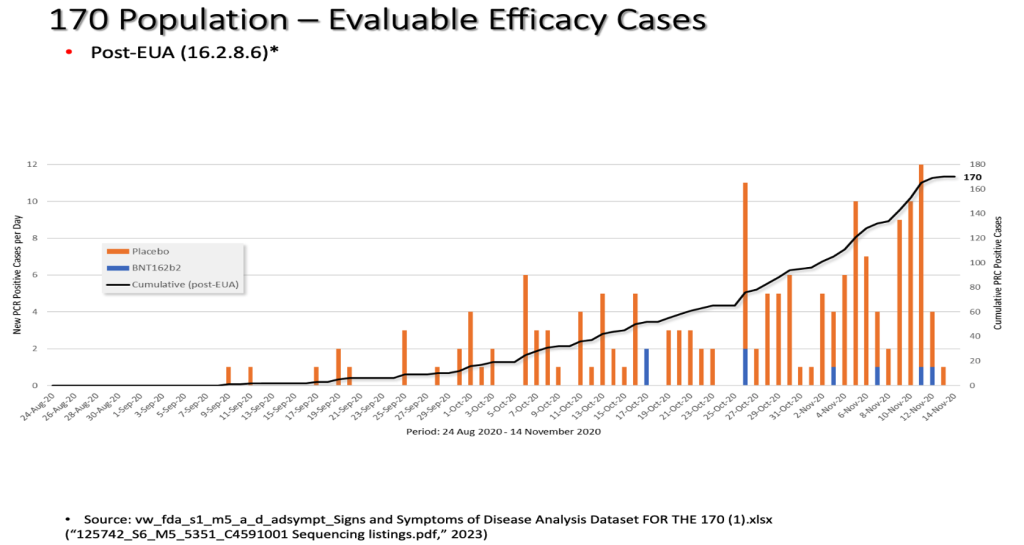
We have also superimposed the rate of accrual of the diagnosis of the 170 population comparing the two differing documents which were finalized at different times about six to seven months apart.
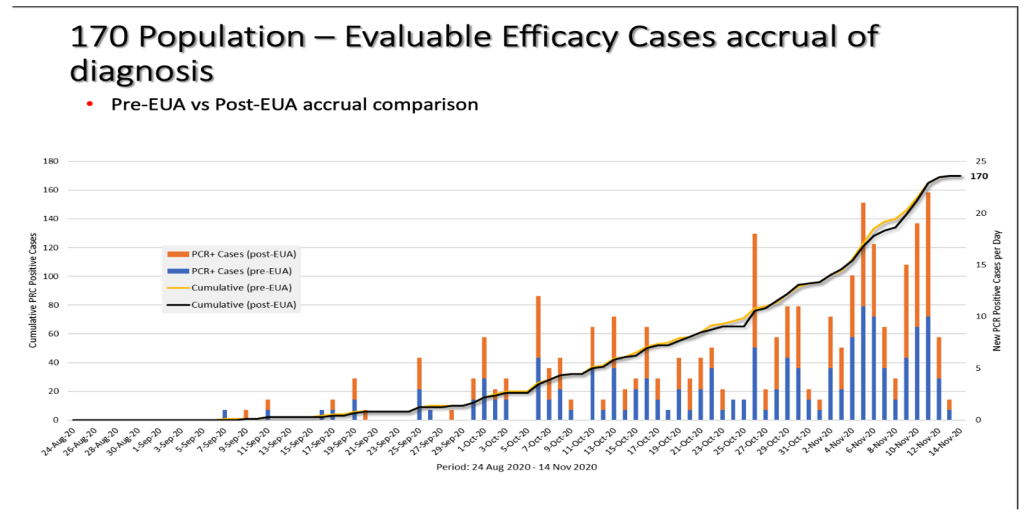
As one can see, the threshold number of efficacy cases is reached around the same time regardless.
Was Pfizer publicly forthright about the time required to accrue the 170 patients?
Now, let us superimpose the two graphs of accrual, the derived one (in red) based on Pfizer’s published documentation, which we introduced earlier, versus the actual accrual (in black) that has been based on the Pfizer document finalized in June 2021 (https://www.phmpt.org/wp-content/uploads/2023/01/125742_S6_M5_5351_c4591001-sequencing-listings.pdf).
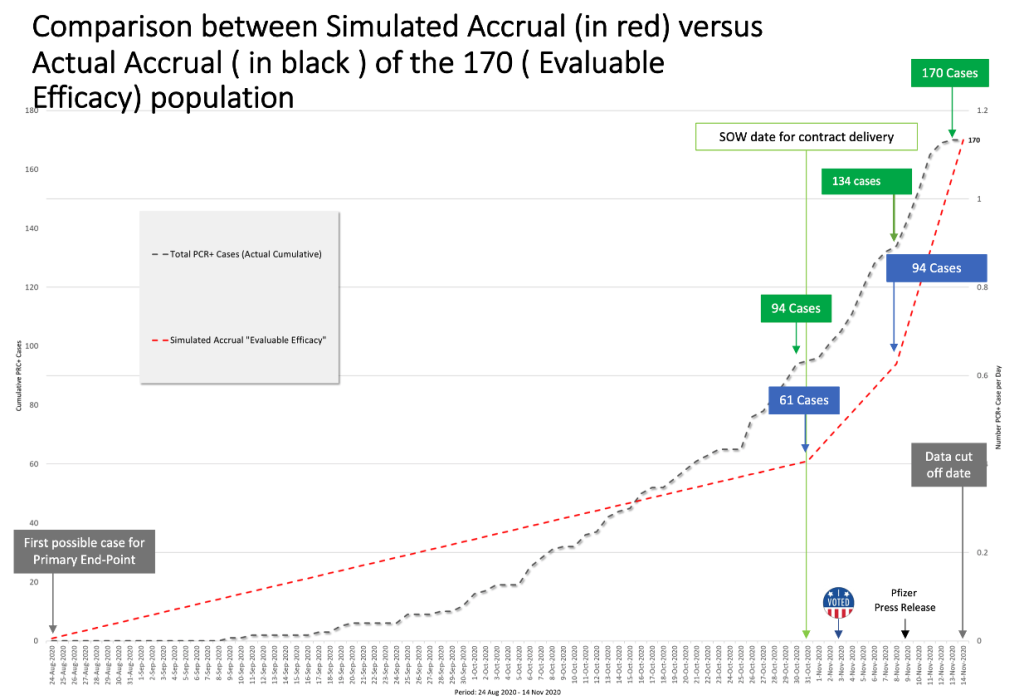
Slide by Ed Clark and Jeyanthi Kunadhasan
Comparing the derived rate of accrual with the actual accrual, we can see a curious discrepancy in the number of evaluable cases diagnosed. Pfizer had accumulated at least 94 cases by October 31, 2020, but it delayed this announcement until a week later on November 9, 2020. What caused this delayed announcement? As highlighted earlier in the SOW (https://www.hhs.gov/sites/default/files/pfizer-inc-covid-19-vaccine-contract.pdf, p. 14), the U.S. government would have had to have been informed of any issue that “…would be reasonably expected to materially change the anticipated schedule by one week or more.” October 31 to November 9 equates to 10 total days, more than the one week stated in the SOW, yet there is no evidence that the U.S. government was informed of this schedule change.
The rate of accumulation of the actual 170 cases in no way resembles what the public was led to believe by Pfizer and legacy media. In October 2020, the rate of accrual of cases is approximately 2.8 cases per day, which is not surprising given that more than half of the trial population had already been recruited by then, thus increasing one’s likelihood of success given the larger pool of participants. By November 8, 2020, there were about 134 COVID cases diagnosed. Why did Pfizer only admit to 94 diagnosed cases as of that date? Even accounting for a few days to evaluate cases, why is there a discrepancy of 40 cases?
The timing of the interim analyses
Remember that the SOW aspired for an FDA-approved or FDA-authorized vaccine by October 31, 2020. In his book Moonshot, Albert Bourla talks about his intention to have a vaccine by October 2020. (Bourla, Albert. (2022). Moonshot. Harper Business, p. 42.) Let us familiarize ourselves with the amendment talked about in the SOW as well as Protocol Amendment 4, the plan for interim analysis of evaluable cases.
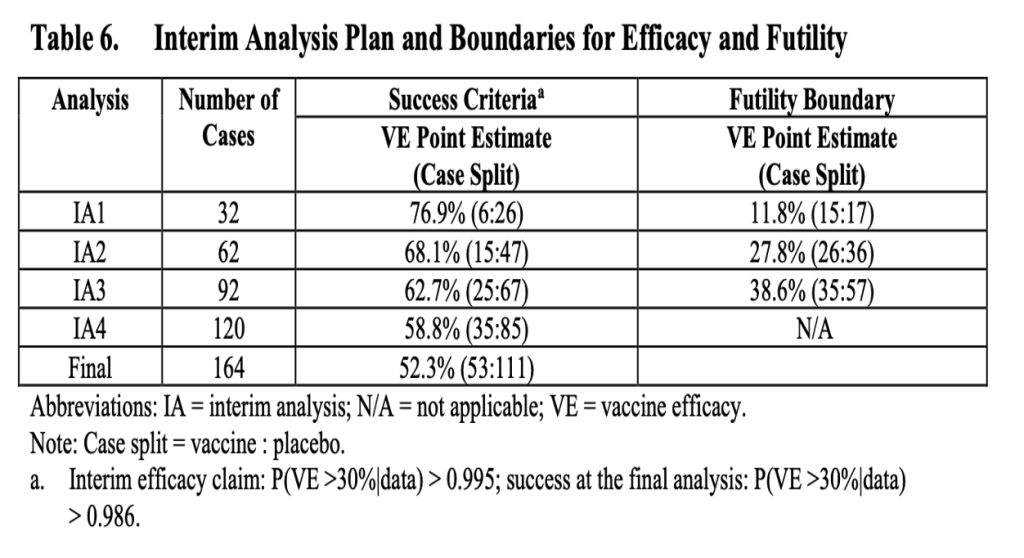
(125742_S1_M5_5351_c4591001-interim-mth6-protocol.pdf. (n.d.). Retrieved from https://phmpt.org/wp-content/uploads/2022/03/125742_S1_M5_5351_c4591001-interim-mth6-protocol.pdf p. 1793.)
Realistically, a vaccine could have only been approved by October 31, 2020, if the first interim analysis had been done at the accrual of 32 cases. This goalpost changed when individuals from the FDA’s Office of Vaccines Research and Review (OVRR), Center for Biologics Evaluation and Research (CBER) penned an article in the New England Journal of Medicine on October 16, 2020, outlining a new requirement that data from Phase 3 studies to support an EUA, which may result from a protocol-specified interim analysis, include a median follow-up duration of at least two months of safety data after completion of the full vaccination regimen. (Krause, P. R., & Gruber, M. F. (2020). Emergency Use Authorization of Covid Vaccines — Safety and Efficacy Follow-up Considerations. The New England Journal of Medicine, 383(19), e107. https://doi.org/10.1056/nejmp2031373.)
The newly imposed safety criteria could only have been fulfilled in the third week of November 2020, as was outlined in Pfizer’s press release (“Pfizer and BioNTech Announce Vaccine Candidate Against COVID-19 Achieved Success in First Interim Analysis from Phase 3 Study.” (n.d.). Retrieved June 19, 2023, from https://www.businesswire.com/news/home/20201109005539/en/) and the EUA documentation (Gruber, M. F. (December 11, 2020). EUA Decision Memorandum. Retrieved from https://www.fda.gov/media/144416/download,) section 4.2.2, p. 17). This is because half of the trial population had been recruited only on September 1, 2020. Pfizer only could have completed the stated vaccination regime for those recruited on September 1, 2020, 21 days later on September 22, 2020. Two months after September 22 is the third week of November, not November 9, 2020, when the interim analysis results were announced. Pfizer was allowed to circumvent the new requirement for a median follow-up of at least two months of safety data after completion of the full vaccination regimen. If the requirement was not going to be followed, why did Pfizer not announce its efficacy findings when 62 cases were accrued, immediately after October 22, 2020, as was promised in the ninth protocol amendment?
We have analyzed when the interim analyses could have been done, as well as estimated vaccine efficacy, according to the dates when enough cases were diagnosed from the 170-patient population.
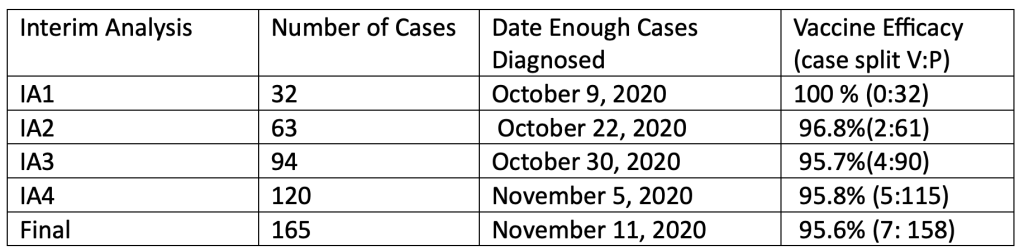
Accrual of the 170-patient population Pfizer documents in intervals where the interim analyses could have been done. (125742_S6_M5_5351_C4591001 Sequencing listings.pdf, finalized June 2, 2021, https://www.phmpt.org/wp-content/uploads/2023/01/125742_S6_M5_5351_c4591001-sequencing-listings.pdf.)
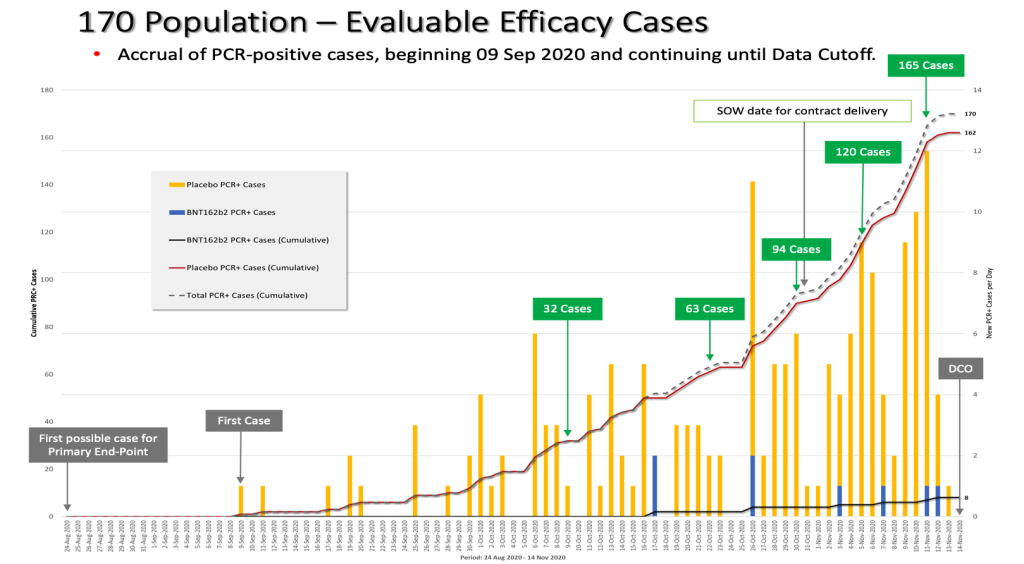
Slide by Ed Clark and J. Kunadhasan
Days’ difference between onset of symptoms and a positive central lab test
In doing this analysis, we have depended on the published Pfizer PDF documents that were released under court order by the FDA. We also have access to the Pfizer XPT database files released by the FDA, which are closer to the raw data Pfizer employees or contractors originally entered. As such, we can double-check the dates published in PDFs for dates the tests were taken and results returned, as well as when symptoms were first reported by patients. The ability to analyze the dates of the positive NAAT (nasal swab) test results from the XPT database was not possible until the January 3, 2023, release of Pfizer data by the FDA, which provided the subject-level .XPT files that contained those data. The data were included in the FDA-CBER-2021-5683-0663135-0671344-125742_S1_M5_c4591001-A-D-adsympt.XPT file (SAS export file for Signs and Symptoms of Disease Analysis Dataset, https://phmpt.org/wp-content/uploads/2023/01/FDA-CBER-2021-5683-0663135-0671344-125742_S1_M5_c4591001-A-D-adsympt.zip). This file was uploaded to the Abstractor website (https://vaccines.shinyapps.io/abstractor/) shortly after it was available and, thus, allowed access to researchers. Additional cross-checks were done with another file delivered in February 2023: FDA-CBER-2021-5683-0710069-0763793-125742_S1_M5_c4591001-A-D-adc19ef.XPT (COVID-19 Efficacy Analysis Dataset, https://pdata0916.s3.us-east-2.amazonaws.com/pdocs/020123/FDA-CBER-2021-5683-0710069-0763793-125742_S1_M5_c4591001-A-D-adc19ef.zip).
The endpoint in this trial was having a symptomatic COVID infection. This was classified as an “illness visit” in the protocol with patients optimally coming in or having a telehealth consultation within three days of potential illness. (125742_S1_M5_5351_c4591001 interim mth6 lab measurements sensitive.pdf. (2022, July 1). Retrieved from https://www.phmpt.org/wp-content/uploads/2022/07/125742_S1_M5_5351_c4591001-interim-mth6-lab-measurements-sensitive.pdf, p. 1629.)
We analyzed the number of days’ difference between the patient reporting symptoms and the central lab test of the nasal swab returning a positive test result. This table shows the number of days’ difference between the Analysis Date for Test (ADT) – i.e., the date the central lab test result was recorded – minus the start date of first symptoms as found in the XPT database files.
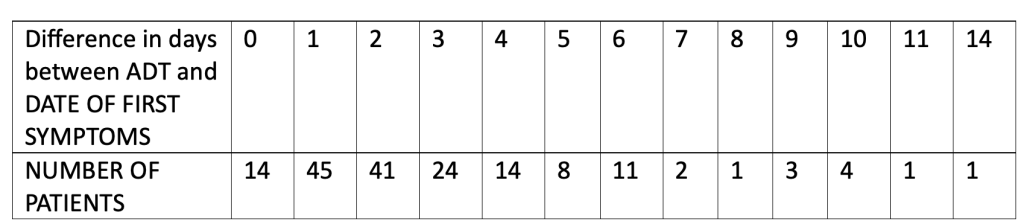
Note that the total equals 169 patients since patient 10911203 did not have a central lab test performed.
ADT (Analysis Date for Test) for RTCOV2NS, CEPHEID RT-PCR ASSAY FOR SARS-COV-2
Most patients, 124 (72.9%), had a confirmatory central lab PCR test for COVID-19 between zero and three days of onset of symptoms, as was optimally suggested for in the protocol. Fourteen patients were able to have a confirmatory nasal swab central lab test, presumably done in the Pearl River facility in New York, on the very day their symptoms started.
So, for 14 people, scattered around the U.S. as well as Argentina, the number of days between the start of symptoms and a central lab test, most likely done in New York state, record of the related nasal swab was zero days. Nine of the 14 patients reported start of symptoms between November 5, 2020, and November 12, 2020. The patient from Argentina (subject ID 12311560) started having COVID symptoms on November 12, 2020, and the confirmatory positive nasal swab was able to be processed in New York that same day. Note that the flight from Buenos Aires to New York City metro area airports is approximately 11 hours. Then, add to that the time needed clear customs with a contagious biological specimen plus the approximate hour courier drive to Pfizer’s lab in Pearl River. This test seems to have, indeed, been completed at Pfizer’s “speed of science” (https://www.youtube.com/watch?v=00VuhThe9Tg&ab_channel=Tipi). Please note, we do not have access to data such as when the time sample was received by a lab, the time the sample was processed, or when it was in the hands of the courier. We have access to when the data was recorded positive by the presumed central laboratory.
For those who had four or more days, up to a total of 14 days, between the onset of symptoms and a confirmatory central lab positive test of infection, 34 of the 45 (75.6%) had onset of symptoms between October 1, 2020, and November 3, 2020. Is there utility in attempting to diagnose a viral respiratory illness up to 14 days after the onset of symptoms? (Pretorius, M., & Venter, M. (2017). Diagnosis of Viral Infections. Viral Infections in Children, Volume I, 151–182. https://doi.org/10.1007/978-3-319-54033-7_6)
We would also like to highlight one of the patients, Subject 11231256, who in the .XPT files is listed as having symptoms from the October 24, 2020. However, in the PDFs and interim narrative files has the start of symptoms listed for Subject 11231256 is October 28, 2020. This patient has a nine-day difference between start of symptoms and having a positive test showing COVID-19 infection returned in the central lab. It would have shown up as only five days, a difference of four days, if taking data from the published PDF files without access to the XPT data. Which date is the correct date? Why does Pfizer have different dates in different documents for the same event and the same Subject?
Looking at this data as well, the accrual of the cases could have happened much earlier if patients had been able to have a central confirmatory test of infection closer to their onset of symptoms, as suggested in the protocol. Had swab results been processed in that way, more test results would have been available prior to the 2020 U.S. presidential election rather than after it.
In the study protocol, it was stated that for “…operational reasons, the interim analysis planned at accrual of 32 cases was removed.” (125742_S1_M5_5351_c4591001-interim-mth6-protocol.pdf. Retrieved from https://phmpt.org/wp-content/uploads/2022/03/125742_S1_M5_5351_c4591001-interim-mth6-protocol.pdf, p. 935.) It has never been explained clearly what exactly these “operational reasons” were. This change was formalized only on October 29, 2020, 13 days after the article about the new requirement of a median follow-up duration of at least two months of safety data after completion of the full vaccination regimen appeared in the NEJM on October 16, 2020. (Krause, P. R., & Gruber, M. F. (2020). Emergency Use Authorization of Covid Vaccines — Safety and Efficacy Follow-up Considerations. The New England Journal of Medicine, 383(19), e107. https://doi.org/10.1056/nejmp2031373.)
There was a discussion between a prominent cardiologist at the Scripps Research Institute, Eric Topol, and the then FDA Commissioner, Steven Hahn, on October 9, 2020, wherein Steven Hahn assured that any approval for the vaccine would be based on “science, not political convenience.” (“FDA’s Hahn to Topol: Decisions Will Be Made Based on Science Alone.” (October 9, 2020). Retrieved from https://www.medscape.com/viewarticle/938705.) (Kunadhasan, Jeyanthi. (March 24, 2023). the powerful politics of Covid vaccines. Spectator.com.au. Retrieved from https://www.spectator.com.au) Was there any peeking into the data then that precipitated the conversation? This is especially important as, officially, the first interim analysis was only admitted to having been completed on November 8, 2020. (https://www.pfizer.com/news/press-release/press-release-detail/pfizer-and-biontech-announce-vaccine-candidate-against) Have any questions been asked as to why Dr. Topol and Commissioner Hahn chose the seemingly fortuitous October 9 date to have such a discussion?
Eric Topol, the author of an open letter [Topol, E., MD. (August 31, 2020). “Dear Commissioner Hahn: Tell the Truth or Resign An Open Letter to Stephen Hahn, MD, From Eric Topol, MD.” Retrieved from https://www.medscape.com/viewarticle/936611] to Stephen Hahn on August 31, 2020, stated in his letter [https://www.medscape.com/viewarticle/936611#vp_3] stated, “…you will not, under any condition, authorize a SARS-CoV-2 vaccine approval before the full Phase 3 completion and read-out of a program.” He then posted this on Twitter two days after the discussion with Commissioner Stephen Hahn.
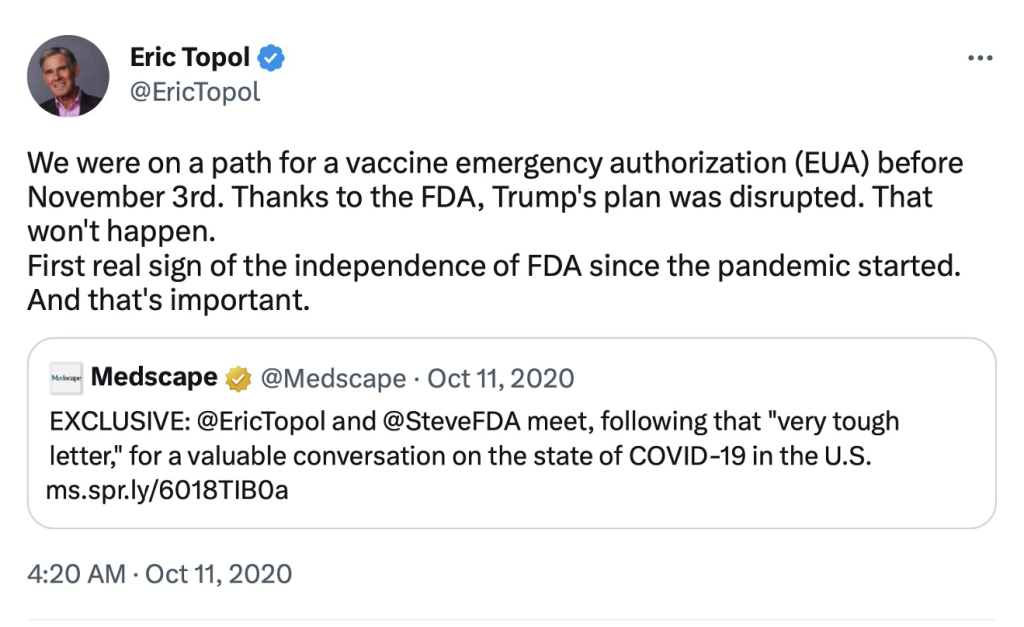
If analyzed at accrual of 32 cases on October 9, 2020, the vaccine would have been 100% efficacious, as there had been no vaccinated patients diagnosed yet with COVID-19. If COVID-19 was such an emergency and this medication allegedly so lifesaving, what was the ethical justification for keeping this news from the public for another month? As we have already elucidated, the “…new safety requirement of median 2 month follow up for safety data…“ was not yet fulfilled when the announcement of the first interim analysis was made on November 9, 2020. (Krause, P. R., & Gruber, M. F. (2020). Emergency Use Authorization of Covid Vaccines — Safety and Efficacy Follow-up Considerations. The New England Journal of Medicine, 383(19), e107. https://doi.org/10.1056/nejmp2031373) If 32 cases were removed due to “operational reasons,” why not do the next interim analysis at accrual of more than 62 cases, which would have been immediately after October 22, 2020? This is extremely important, as the terms of the SOW contract could have been fulfilled if the first interim analysis had been done around October 9, 2020, when 32 cases were accrued.
In summary,
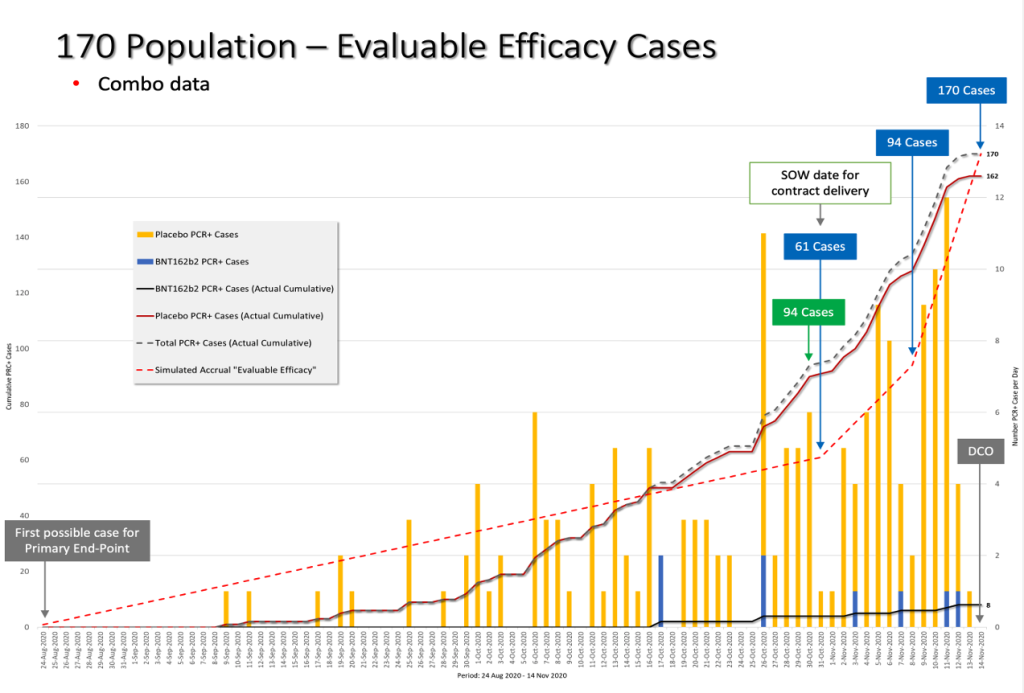
Slide by Ed Clark and Jeyanthi Kunadhasan
The green box highlights when 94 cases were actually accrued versus the blue box, when the official announcement was made by Pfizer after blowing past the contract delivery date of October 31, 2020.
As we can see, on the date of Protocol Amendment 9, October 29, 2020, there were already more than 62 evaluable efficacy cases diagnosed as required in that exact amendment. We have already highlighted previously that the dosing interval between Dose 1 and Dose 2 in this trial was changed WITHOUT a formal protocol amendment, so this makes us take more seriously any protocol amendment that has been made. (Kunadhasan, J, Clark, E., & Flowers, C. (October 21, 2022). “Report 42: Pfizer’s EUA Granted Based on Fewer Than 0.4% of Clinical Trial Participants. FDA Ignored Disqualifying Protocol Deviations to Grant EUA.” Retrieved from https://dailyclout.io/report-41-the-170-clinical-trial-participants-who-changed-the-world-pfizer-ignored-protocol-deviations-to-obtain-emergency-use-authorization-for-its-covid-19-mrna-vaccine/)
Is it reasonable to ask if the number of evaluable cases diagnosed – i.e., the pivotal efficacy data – was a deliverable closely monitored by all parties to the SOW? After all, the SOW mentioned this “pivotal efficacy portion.” (https://www.hhs.gov/sites/default/files/pfizer-inc-covid-19-vaccine-contract.pdf, pp. 6-7.) For the contract to have been extended in good faith, what was the number of evaluable efficacy cases admitted to by Pfizer when the contract had to be modified after October 31, 2020? The late modification of the trial protocol on October 29, 2020, stating that the interim analysis would be done when there were at least 62 cases diagnosed, leads us to the obvious question — was the number of evaluable efficacy cases fraudulently underrepresented as to when they been diagnosed?
Very few people will know the answer to that question, and much of the SOW and the subsequent contract modification remain redacted. (https://www.hhs.gov/sites/default/files/pfizer-inc-covid-19-vaccine-contract.pdf) So, we are unable to answer this simple but vitally important question. We hope by raising awareness regarding this issue, so the public can get answers.
Data Methods for Determining the Timing of the 170 Patients’ Data
The ability to analyze the dates of the positive NAAT test results was not possible until the January 3, 2023, data production which provided the subject-level .XPT data files that contained this data. The data were included in the FDA-CBER-2021-5683-0663135-0671344-125742_S1_M5_c4591001-A-D-adsympt.XPT (SAS export file for Signs and Symptoms of Disease Analysis Dataset). (https://phmpt.org/wp-content/uploads/2023/01/FDA-CBER-2021-5683-0663135-0671344-125742_S1_M5_c4591001-A-D-adsympt.zip) This file was uploaded to the Abstractor website (https://vaccines.shinyapps.io/abstractor/) shortly after it was available, which allowed access to researchers.
The fields of interest in this data set are:
USUBJID – Unique Subject Identifier
PARAMCD – Parameter Code
RTCOV2NS – CEPHEID RT-PCR ASSAY FOR SARS-COV-2 (This is the test that was run in Pearl River, New York)
SARSCOV2 – SEVERE ACUTE RESP SYNDROME CORONAVIRUS 2 (This is the local test.)
PARCAT1 – Parameter Category 1
AVALC – Analysis Value (C)
This provides the results of the analysis.
POS (Shows that there was a positive test result for the test in question.)
ADT – Analysis Date for Test
Date entered for the test result
ASDT – Analysis Start Date
Date that first symptoms were entered
Steps:
- Get the USUBJID for the 170 patients.
- Select the data from the Signs and Symptoms of Disease Analysis Dataset for the 170 patients.
- Verify the date of the first sign and symptom of disease (PARCAT1 = SIGNS AND SYMPTOMS OF DISEASE, AVALC=Y, ASDT is the resulting date for the first sign and symptom of disease). This should match the date in the PDF 16.2.8.6. (125742_S6_M5_5351_c4591001 sequencing listings.pdf. (2023, January 3). Retrieved from https://www.phmpt.org/wp-content/uploads/2023/01/125742_S6_M5_5351_c4591001-sequencing-listings.pdf.)
- Verify that the date of the New York validation matches the “Swab Date” listed in 16.2.8.6 125742_S6_M5_5351_c4591001 sequencing listings.pdf. (2023, January 3). (Retrieved from https://www.phmpt.org/wp-content/uploads/2023/01/125742_S6_M5_5351_c4591001-sequencing-listings.pdf.)
- . The New York validation date is determined from the Symptoms of Disease Analysis (PARAMCD=RTCOV2NS, PARCAT1=VIROLOGY, AVALC=POS), and ADT is the resulting date when the test result said that there is a POS test result in New York.
Additional cross-checks were done against another file produced by the FDA in February 2023 (FDA-CBER-2021-5683-0710069-0763793-125742_S1_M5_c4591001-A-D-adc19ef.XPT, COVID-19 Efficacy Analysis Dataset, https://pdata0916.s3.us-east-2.amazonaws.com/pdocs/020123/FDA-CBER-2021-5683-0710069-0763793-125742_S1_M5_c4591001-A-D-adc19ef.zip).
170 Efficacy Population Analysis
(Download: 170 Efficacy Population Analysis in Excel)
Here’s a link to the full report:











Vaccine News
Tennessee Woman Fired for Refusing Employer’s COVID-19 Vaccine Mandate Wins Almost $700K
Vaccine News
Kansas Sues Pfizer Over Alleged Consumer Protection Violations Related to COVID-19 Vaccine

Vaccine News
mRNA Covid Vaccine Aids Development of Cancer, New Study Finds

Bipartisan House Task Force to Investigate Assassination Attempt on Donald Trump

Justice Department Finds Transcripts They Previously Denied Existence of in Biden Classified Material Investigation

Leaked Emails Expose BBC’s Unverified Reporting and Political Bias

2020 Georgia Fraud Exposed
Tennessee Woman Fired for Refusing Employer’s COVID-19 Vaccine Mandate Wins Almost $700K

U.S. Military Has Started Recalling Retirees Due to Recruiting Crisis
-

 Vaccine News3 weeks ago
Vaccine News3 weeks agoTennessee Woman Fired for Refusing Employer’s COVID-19 Vaccine Mandate Wins Almost $700K
-
Trending2 weeks ago
Elon Musk Backs Voter Bill Aimed at Providing Proof of U.S. Citizenship to Vote, Labels Opponents as “Traitors”
-
2024 Race3 days ago
Hillary Clinton Planning Run for 2024 Presidency, According to Trump Campaign
-
2024 Race4 days ago
DNC to Proceed with Plan to Confirm Joe Biden as Presidential Nominee
-
Politics2 weeks ago
Democrats Block SAVE Act in Senate, Allowing Potential for Illegal Immigrant Voting
-
Trending2 weeks ago
Vending Machines are Now Selling Bullets at Grocery Stores
-
WEF3 weeks ago
Former World Economic Forum Staffers Accuse Founder Klaus Schwab of Sexual Harassment
-
Trending2 weeks ago
Supreme Court Ruling Alters January 6 Charges: From Insurrection to Trespassing












You must be logged in to post a comment Login